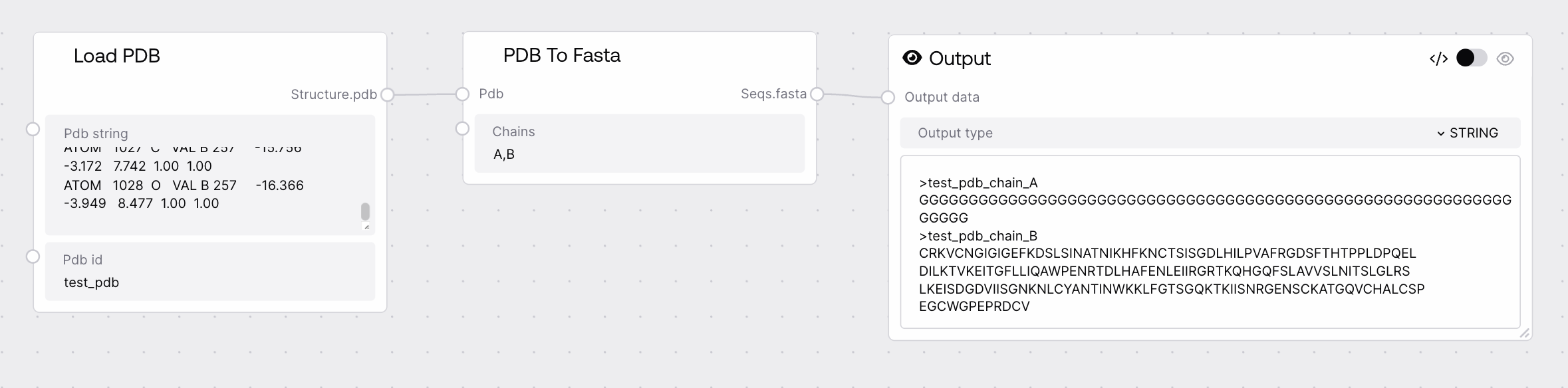
PDB To Fasta converts uploaded PDB structures into FASTA-formatted amino acid sequences. It parses ATOM records in the PDB, groups them by chain, and reconstructs the residue sequence using standard amino acid codes. This node is typically used to derive clean, textual protein sequences from structural models so they can be used in sequence-based workflows or inspected independently.
Use PDB To Fasta when you have 3D protein structures (e.g., from AlphaFold, RFdiffusion, docking tools, or experimental PDB files) and you need their corresponding amino acid sequences in FASTA form. It usually appears downstream of structure-loading or generation nodes such as LoadPDBNode, Alphafold-related nodes, or docking/evaluation nodes, and upstream of sequence-based analysis or design nodes such as LoadFastaNode consumers, MSA generation, immunogenicity prediction, or sequence comparison tools. In a typical workflow, you might: (1) load or generate one or more PDB structures with LoadPDBNode or a modeling node, (2) convert them with PDB To Fasta to obtain per-chain sequences, (3) feed those sequences into MSA search, ProteinMPNN, immunogenicity prediction, or custom analysis nodes. PDB To Fasta pairs naturally with FastaCombinerNode for merging outputs into a single multi-entry FASTA, and with PDBChainExtractorNode when you first want to isolate specific chains before sequence extraction. Prefer using this node whenever you want the sequence actually represented in a PDB, instead of relying on external or assumed sequences.
Dictionary of PDB structures to convert. Expected format is {pdb_id: pdb_content} where pdb_content is a valid PDB text string containing ATOM records. Each entry may contain multiple chains; the node will derive sequences per chain. PDB content must be non-empty and correctly formatted with standard residue and chain fields.
{"1ABC_model": "HEADER TEST PDB\nATOM 1 N MET A 1 ...\nATOM 2 CA MET A 1 ...\nATOM 3 N GLY B 1 ...\nEND"}
FASTA-formatted sequences derived from the input PDBs, typically as a dictionary {sequence_id: fasta_string}. Each sequence_id may encode both the original PDB identifier and the chain (e.g., 1ABC_A, 1ABC_B). Each fasta_string is a standard FASTA entry with a header line and a contiguous one-letter amino acid sequence, suitable for downstream sequence-based tools.
{"1ABC_A": ">1ABC_A chain A derived from PDB\nMKTFFVLALVLSGALA\n", "1ABC_B": ">1ABC_B chain B derived from PDB\nGGHGLYPNVTRW"}
Performance: Parsing is line-based over ATOM records; for very large PDBs or many input structures this can be moderately CPU-intensive but is generally fast for typical protein sizes.
Limitations: Only standard PDB ATOM/HETATM records are supported; malformed PDBs or exotic residue naming may lead to missing or unknown residues in the output sequence.
Limitations: Non-standard amino acids and ligands are usually skipped or mapped conservatively, so they may not appear faithfully in the resulting sequence.
Behavior: Sequences are usually grouped by chain; if an input PDB contains multiple models or chains, you should expect multiple FASTA entries per PDB ID.
Behavior: Empty or structurally invalid PDB entries will typically cause validation errors rather than silently producing empty sequences.
No sequence found / empty FASTA output: Check that the input PDB text actually contains ATOM records for the intended chains and is passed as a dictionary {id: content}; loading a raw string or an empty file will produce no sequences.
KeyError or type error on input: This usually indicates that the node received something other than a PDB-typed dictionary (e.g., a plain string or list). Ensure you connect a PDB output (e.g., from LoadPDBNode or PDBCombinerNode) directly to PDB To Fasta.
Unexpected or truncated sequence length: If your PDB is missing residues or contains only partial chains, the resulting FASTA will reflect that. Verify the structural completeness of the input PDB and consider using PdbFixerNode or PDBChainExtractorNode beforehand if you need a specific region.
Mixed or unexpected chain labeling in headers: Sequence IDs combine the original PDB identifier and chain; if upstream nodes renamed chains (e.g., PDBCombinerNode with prefixing), the FASTA headers will reflect those modified chain IDs. Inspect upstream settings if headers look unfamiliar.