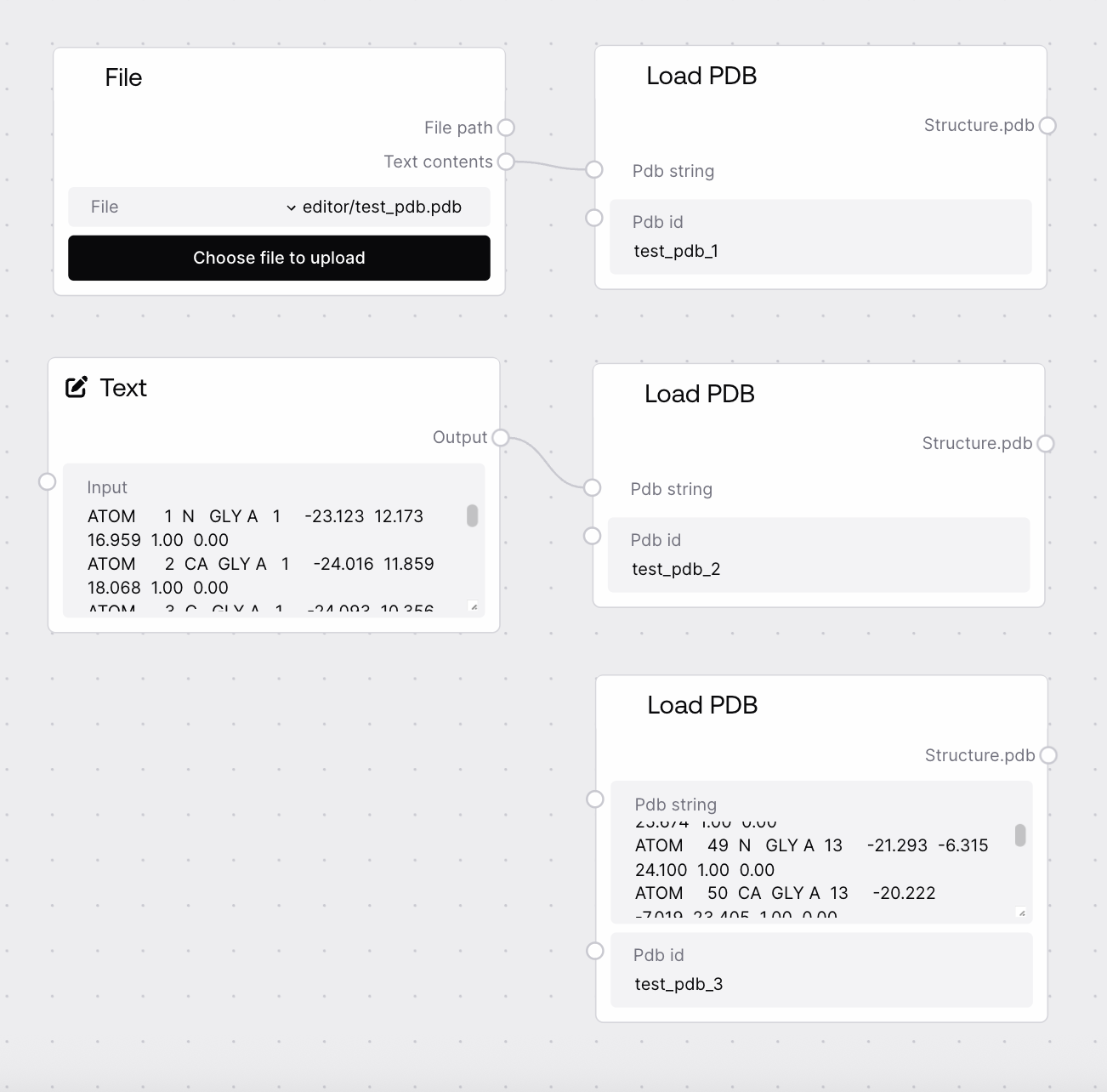
Load PDB takes a raw PDB-format text string and associates it with a user-specified identifier, returning a PDB-typed dictionary. The output has the form {pdb_id: pdb_string} and includes Salt workflow metadata from its base class. This provides a clean, standardized entry point for protein structures in biotech pipelines.
Use this node at the start of any workflow that needs to operate on protein structures in PDB format. Typical use cases include loading a PDB copied from the Protein Data Bank, importing a PDB produced by an external modeling tool, or defining multiple structures with separate Load PDB nodes and combining them later. After loading, connect its output to nodes such as "PDB Visualization" (for web-based viewing), "PDB To Fasta" (for sequence extraction), "PDB Chain Extractor" (to isolate chains A, B, etc.), or "PDB Fixer" (to replace a chain sequence using a FASTA input). When handling multiple structures, give each Load PDB node a unique pdb_id, then merge them using "Batch PDB" or "PDB Combiner" before exporting with "Save To Zip" or "Save To Bucket". The node does not validate the PDB format itself, so you should only provide well-formed PDB strings.
Full PDB content as plain text. This should be the exact text you would find in a .pdb file, typically including lines like HEADER, TITLE, ATOM, HETATM, and END. The node does not parse or validate the format; malformed or non-PDB text will be passed through but may fail later in nodes that parse structures.
HEADER OXYGEN TRANSPORT 07-JUL-97 1A3N ATOM 1 N VAL A 1 11.104 13.207 5.678 1.00 20.00 N ATOM 2 CA VAL A 1 12.560 13.410 5.432 1.00 20.00 C ATOM 3 C VAL A 1 13.014 14.861 5.741 1.00 20.00 C END
pdb_id
True
STRING
Identifier that will be used as the key in the output PDB dictionary. It should be unique for each structure within a workflow, especially if you plan to combine outputs from multiple Load PDB nodes. If you also load matching FASTA or A3M data, use the same ID there to keep records aligned.
A PDB-typed dictionary mapping pdb_id to the provided PDB string, augmented with internal Salt metadata. Downstream biotech nodes expect this dictionary structure when consuming PDB inputs, allowing them to handle both single structures and batches in a consistent way.
{'1a3n_chainA': 'HEADER OXYGEN TRANSPORT 07-JUL-97 1A3N\nATOM 1 N VAL A 1 11.104 13.207 5.678 1.00 20.00 N\nATOM 2 CA VAL A 1 12.560 13.410 5.432 1.00 20.00 C\nATOM 3 C VAL A 1 13.014 14.861 5.741 1.00 20.00 C\nEND\n'}
Performance: The node simply wraps the provided text in a PDB-typed container and adds metadata; it performs no structural parsing, so runtime is minimal even for large PDB files.
Limitations: There is no built-in validation that pdb_string is valid PDB; errors from bad formatting, missing fields, or truncated files will typically appear only in downstream nodes that parse the structure.
Behavior: The output always contains exactly one entry keyed by pdb_id. If downstream combiner nodes receive multiple dictionaries with the same key, they will raise errors about duplicate IDs.
Behavior: The Salt metadata added by this node is used by exporter nodes (like Save To Zip and Save To Bucket) for organizing folder and file names when writing structures to disk or cloud storage.
Common Error 1: A combiner node reports a message about a repeated PDB id (for example, that a given ID appears twice). This indicates that two Load PDB nodes used the same pdb_id. Fix the issue by assigning unique pdb_id values to each structure.
Common Error 2: A downstream node complains that the PDB input is empty or not a dictionary. This usually happens if you connect a node to a raw string instead of the Load PDB output, or if the workflow branch is not executed. Ensure the consumer node is wired to the "structure.pdb" output and that Load PDB is part of the executed path.
Common Error 3: Nodes that inspect chains or sequences (such as PDB To Fasta or PDB Chain Extractor) fail with messages about missing chains or atoms. In that case, verify that pdb_string contains well-formed ATOM/HETATM records, correct chain IDs, and a complete structure; Load PDB itself will not reject malformed input.